


-
MICROCEFALIA
La microcefalia se define como un perímetro cefálico (PC) menor de 2 desviaciones estándar (DE) para la media de edad, sexo y edad gestacional. No se trata de un diagnóstico, sino de un signo que puede estar asociado a diversas patologías o ser una variante de la normalidad.
Encontramos tres definiciones, aunque sujetas a cierta controversia:
-
Microcefalia límite: PC 2-3 DE por debajo de la media.
-
Microcefalia moderada: PC 3-5 DE por debajo de la media.
-
Microcefalia grave: PC >5 DE por debajo de la media.
Hay que tener en cuenta que la definición de microcefalia límite incluiría un porcentaje de sujetos que se considerarían por lo demás sanos.
También pueden considerarse microcefalia aquellos casos en los que el perímetro cefálico atraviesa dos o más líneas principales de percentil (95, 75, 50, 25,10) en sentido descendente en dos controles correlativos de salud.
Hay que realizar además una distinción entre microcefalia y microencefalia, que hace referencia a un cerebro de menor tamaño de lo normal. Si bien un alto porcentaje de casos de microcefalia se asocian a microencefalia, debido a que el tamaño del cráneo está determinado en buena parte por el crecimiento encefálico, puede existir microencefalia en pacientes con perímetro cefálico normal.
Dentro de la patogenia de la microcefalia destacan dos posibles mecanismos principales:
-
Alteraciones en la neurogénesis, con menor número de neuronas generadas, frecuentemente de tipo congénito.
-
Lesión en un cerebro previamente sano, que disminuye las conexiones sinápticas establecidas, frecuentemente asociado a cuadros adquiridos1-5.
-
-
CLÍNICA DE CUADRO SINDRÓMICO
Existe una gran variedad de cuadros sindrómicos asociados a microcefalia. Destacamos los siguientes, con sus manifestaciones clínicas características:
-
Síndrome de Down: braquicefalia, fisuras palpebrales inclinadas superiormente, pliegues en epicanto, cuello corto, espacio entre el primer y segundo dedo del pie, hipotonía.
-
Trisomía 18: occipucio prominente, diámetro frontal estrecho, hipoplasia de relieve supraorbital, fisuras palpebrales cortas, micrognatia, lesiones cardiacas estructurales.
-
Trisomía 13: holoprosencefalia, sutura sagital amplia, labio leporino y fisura palatina, piel fláccida, pliegue palmar transverso, polidactilia, prominencia posterior del talón, alteraciones cardiacas estructurales.
-
Síndrome de Seckel: retraso en el crecimiento pre y posnatal, micrognatia, asimetría facial, fisuras palpebrales inclinadas hacia abajo, nariz aguileña y prominente, hipoplasia de miembros, espacio entre el primer y el segundo dedo del pie.
-
Síndrome de Smith-Lemli-Opitz: ptosis, punta de la nariz amplia, anteversión de narinas, micrognatia, fisura palatina, defectos cardiacos congénitos, sindactilia de primer y segundo dedos del pie, polidactilia postaxial, hipospadias y criptorquidia.
-
Síndrome de Williams-Beuren: estenosis aórtica supravalvular, hipercalcemia idiopática, congestión periocular, nariz corta y orientada hacia arriba, filtrum largo, boca amplia, labios carnosos.
-
Síndrome de Cornelia de Lange: retraso en el crecimiento pre y posnatal, hirsutismo generalizado, fusión y arqueamiento de cejas, nariz corta y orientada hacia arriba, pestañas largas, pico medial en labio superior, falta de dedos, sindactilia entre el segundo y tercer dedo del pie.
-
Lisencefalia de Miller-Dieker: estrechamiento bitemporal, nariz orientada hacia arriba, mandíbula pequeña, pliegues verticales en la frente, micrognatia, alteraciones genitourinarias.
-
Síndrome de Wolf-Hirschhorn: cardiopatía congénita, pérdida auditiva, glabela prominente, hipertelorismo, puente nasal amplio, nariz aguileña, filtrum corto, labio superior girado hacia abajo.
-
Síndrome de maullido de gato: cara redonda, hipertelorismo, micrognatia, pliegues en epicanto, hipotonía, llanto agudo.
-
Delección 1p36: braquicefalia, fontanela agrandada, barbilla puntiaguda, pérdida de audición, puente nasal planto, labio leporino, fisura palatina, quinto dedo corto.
-
Síndrome Mowat-Wilson: microcefalia pre o posnatal, talla baja, hipertelorismo, coloboma de iris, ojos hundidos, fisuras palpebrales orientadas hacia abajo, orejas ahuecadas, barbilla puntiaguda, crisis convulsivas, hipospadias, enfermedad de Hirshprung, cardiopatía congénita.
-
Síndrome de Rubinstein-Taybi: talla baja posnatal, implantación anterior del pelo baja, maxilar hipoplásico, micrognatia, cejas pobladas, pestañas largas, pulgares anchos, dedos del pie grandes.
-
Síndrome de Aicardi-Goutières: microcefalia congénita, movimientos oculares anormales, hepatoesplenomegalia, calcificaciones cerebrales, trombocitopenia, espasticidad, crisis convulsivas.
Los cuadros sindrómicos son difíciles de diagnosticar fenotípicamente, salvo que dicho fenotipo sea muy claro. El diagnóstico genético es la herramienta más útil en este caso, siendo la neuroimagen otro elemento que puede ser de utilidad, y convendrá realizar derivación a consulta de dismorfología y a las consultas necesarias dependiendo de la afectación que asocie el cuadro. Pueden encontrarse más fenotipos en bases de datos como Online Mendelian Inheritance in Man4,5,8-10.
-
-
SOSPECHA DE CUADRO CONGÉNITO
Entre las principales causas ambientales de microcefalia congénita se encuentran:
-
Infecciones intraútero: fundamentalmente, TORCH:
-
Toxoplasma: fiebre, rash maculopapular, hepatoesplenomegalia, microcefalia, crisis convulsivas, ictericia, trombocitopenia y linfadenopatías generalizadas. La tríada clásica incluye: coriorretinitis, hidrocefalia y calcificaciones intracraneales.
-
Rubeola: presenta, fundamentalmente, hipoacusia, cardiopatías congénitas, microcefalia, cataratas y restricción del crecimiento intrauterino.
-
Citomegalovirus: genera restricción del crecimiento intrauterino, microcefalia, hipoacusia neurosensorial, petequias e ictericia.
-
Virus herpes simple: microcefalia, encefalomalacia quística, hidranencefalia, lesiones cutáneas y daños oculares11.
-
-
Exposición a tóxicos: tanto drogas (alcohol, cocaína, opiáceos…) como ciertos medicamentos (antineoplásicos y antiepilépticos, como carbamazepina, fenitoína, barbitúricos y ácido valproico).
-
Síndrome alcohólico fetal: retraso en el crecimiento pre y posnatal, fisuras palpebrales cortas, filtrum plano, labio superior fino, alteraciones neurocognitivas y comportamentales1,12.
-
-
Evento hipóxico-isquémico intraútero.
-
Patología materna: fenilcetonuria mal controlada, insuficiencia placentaria, malnutrición, anemia grave, otras patologías sistémicas3.
-
Dado que estos casos son ambientales, puede llegarse a su diagnóstico a través de la historia clínica y, en el caso de las infecciones congénitas, pueden realizarse además serologías dirigidas al microorganismo causante. En este caso conviene además realizar una derivación a la consulta de Infectología Pediátrica.
Existen otras causas de microcefalia congénita, asociadas a malformaciones del sistema nervioso central y microcefalias primarias monogénicas5,13,14. Para sospecharlas será necesario avanzar más en el algoritmo.
-
-
ANAMNESIS
Son elementos esenciales en la anamnesis los siguientes:
-
Momento de aparición: una microcefalia congénita es notable al nacimiento e incluso entre las 18 y 22 semanas de edad gestacional.
-
Factores gestacionales: infecciones (TORCH, VIH, sífilis, enterovirus, Zika), sustancias (alcohol, tabaco, marihuana, opioides, cocaína, antineoplásicos, antiepilépticos, tolueno), radiación, isquemia…
-
Factores perinatales: hipoglucemia, hipotiroidismo, hipopituitarismo, prematuridad y alteraciones en ecografía transfontanelar.
-
Daños sobre el sistema nervioso central: anoxia, isquemia, meningitis, traumatismo craneoencefálico.
-
Medidas antropométricas al nacimiento y su trayectoria.
-
Neurodesarrollo, historia de clínica neurológica y antecedente de crisis convulsivas.
-
Historia familiar: microcefalia (y en caso de haberla, la presencia de clínica asociada), patología neurológica y metabólica, consanguineidad1.
-
-
EXPLORACIÓN FÍSICA
Son elementos esenciales en la exploración física:
-
Rasgos dismórficos (teniendo en cuenta que en las primeras evaluaciones pueden ser difíciles de categorizar y que la microcefalia puede distorsionar per se los rasgos).
-
Perímetro cefálico: el perímetro cefálico es una medida antropométrica de importancia capital. Deberá medirse en todos los controles del programa de salud infantil hasta los 3 años y durante todo el seguimiento de niños con clínica neurológica asociada. Requiere una buena técnica y debe tenerse en cuenta que en las primeras 24 horas de vida puede alterarse por cefalohematoma, caput sucedaneum y cambios en la conformación de la cabeza. Se medirá usando una cinta métrica flexible, pero no elástica, la circunferencia que pasa por el occipucio y 1-2 cm por encima de la eminencia ciliar. Debe contrastarse mediante gráficas con el resto de población de la misma edad, sexo y edad gestacional6. Es importante tener en cuenta que el seguimiento del perímetro cefálico tiene una gran importancia, por lo que hay que comprobar que su ritmo de crecimiento es el adecuado. La velocidad de crecimiento del perímetro cefálico suele ser:
-
Primer trimestre de vida: 2 cm/mes.
-
Segundo trimestre: 1 cm/mes.
-
Tercer trimestre: 0-5 cm/mes.
-
El crecimiento de la cabeza estará prácticamente completo a los 4 años de edad7.
-
-
Peso, talla y su trayectoria.
-
Cabeza: forma de la cabeza y palpación de fontanelas. La fontanela anterior cierra en torno a los 10-24 meses de vida. Un cierre precoz debe hacernos pensar en craneosinostosis, hipertiroidismo o hipoparatiroidismo. Un cierre tardío puede asociarse a cuadros sindrómicos o exposición a tóxicos.
-
Ojos: las alteraciones oculares sugieren infección intrauterina o enfermedad metabólica.
-
Línea media: los defectos en la línea media se asocian a holoprosencefalia (por ejemplo, incisivo maxilar único, coana única…).
-
Piel: hay lesiones asociadas a infección intrauterina (petequias, ictericia…) y a procesos metabólicos (rash eccematoso en la fenilcetonuria).
-
Abdomen: las megalias son indicativas de infección intrauterina y de enfermedad metabólica.
-
Exploración neurológica e hitos del desarrollo1,5.
-
-
HISTORIA DE INSULTO O LESIÓN POSNATAL CONOCIDA
Son cuadros adquiridos capaces de generar microcefalia:
-
Eventos hipóxico-isquémicos perinatales.
-
Hipoglucemia, hipotiroidismo o hipopituitarismo perinatales.
-
Meningitis1.
-
-
CUADRO FAMILIAR
La valoración de la influencia de la genética puede hacerse en microcefalias límite sin clínica ni trastornos del neurodesarrollo asociados. Para ello, se utilizará la gráfica de Weaver (Figura 1).
Es necesario para ello calcular la puntuación estándar o Standard Score (SS) del perímetro cefálico del niño y de sus progenitores, usando para ello datos de la tabla de Nellhaus (Tabla 1). Para los padres se usarán los datos correspondientes a 18 años si tienen más de esta edad.
Standard Score (SS) = (Perímetro cefálico – valor de la media) / Desviación estándar.
Una vez obtenidos los tres SS, se utilizará el del niño y la media del de los padres para localizar un punto en la gráfica, que en el caso de encontrarse dentro del área delimitada indicaría que la microcefalia es de origen familiar.
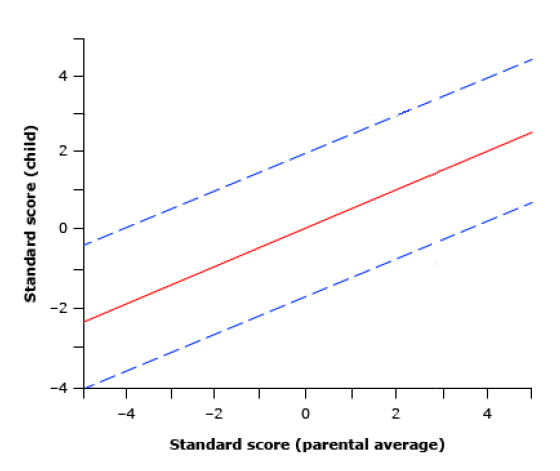
Figura 1. Gráfica de Weaver15.
Tabla 1. Tabla de Nellhaus
Edad
Hombres
Mujeres
Media (cm)
1 DE
Media (cm)
1 DE
Nacimiento
34,74
1,33
34,02
1,22
1 m
37,30
1,30
36,43
1,22
3 m
40,62
1,23
39,71
1,20
6 m
43,76
1,29
42,68
1,38
9 m
45,75
1,28
44,69
1,30
12 m
47,00
1,31
45,81
1,29
18 m
48,31
1,36
47,27
1,36
2 a
49,19
1,39
48,02
1,29
3 a
50,63
1,38
49,25
1,36
4 a
50,91
1,39
50,10
1,37
5 a
51,41
1,37
50,55
1,32
6 a
51,40
1,41
50,52
1,31
7 a
52,24
1,52
51,46
1,35
8 a
52,35
1,40
51,64
1,44
9 a
52,58
1,44
51,87
1,33
10 a
53,16
1,41
52,15
1,50
11 a
53,25
1,53
52,64
1,39
12 a
53,71
1,52
53,01
1,50
13 a
54,14
1,57
53,70
1,37
14 a
54,59
1,30
54,04
1,39
15 a
54,95
1,51
54,39
1,34
16 a
55,37
1,11
54,64
1,16
17 a
55,77
1,32
54,78
1,35
18 a
55,95
1,34
54,94
1,40
Ejemplo: aplicamos la gráfica de Weaver a un niño de 3 meses con un perímetro cefálico de 38 cm (p <1, -2,65 DE). Su madre tiene un perímetro cefálico de 52 cm y su padre de 55 cm.
-
SS niño: (38 cm-40,62 cm) /1,23 cm = -2,13
-
SS madre: (52 cm-54,94 cm) /1,40 cm = -2,1
-
SS padre: (54 cm-55,95 cm) /1,34 cm = -1,45
-
SS medio de los padres: -1,78
Representándolo en la gráfica (Figura 2), observamos que el punto de intersección se encuentra dentro del área, por tanto, podemos decir que la microcefalia es de origen familiar1,15.
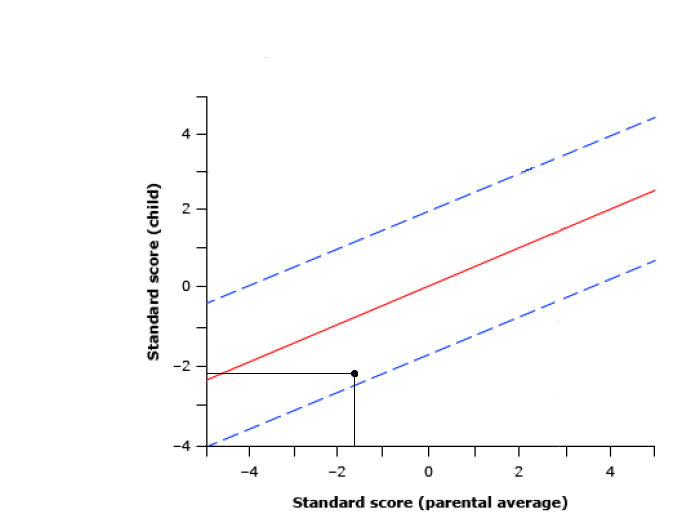
Figura 2. Ejemplo de aplicación de la gráfica de Weaver.
-
-
ESTUDIO ETIOLÓGICO
Las pruebas diagnósticas que son de mayor utilidad son:
-
Pruebas de neuroimagen: están indicadas, principalmente, en caso de alteraciones en el desarrollo y clínica neurológica, pero son técnicas útiles en el estudio de cualquier microcefalia que requiera profundización. La prueba más adecuada es la resonancia magnética nuclear (RMN), que permitirá encontrar malformaciones asociadas a la microcefalia, como la holoprosencefalia, defectos del tubo neural o la lisencefalia, entre otros. Esto facilitará la caracterización del cuadro y la obtención del diagnóstico. La tomografía computarizada (TC) es más útil a la hora de estudiar estructuras óseas (tener en cuenta en la craneosinostosis) y calcificaciones.
-
Estudio metabólico: si se sospecha patología de índole metabólico, puede ser útil en cuadros pre y posnatales
-
Estudio genético: no solo orientado a cuadros sindrómicos, sino también a microcefalias primarias monogénicas, que se caracterizan por presentar un encéfalo con pocas alteraciones a nivel estructural y que pueden presentar clínica poco llamativa, casi siempre con retraso cognitivo más o menos importante y que pueden manifestarse de manera congénita.
-
Valoración oftalmológica y auditiva: es útil en cuadros de infecciones congénitas y sindrómicos1,2,5.
-